MurciaSalud
CSUR ERN
Información para pacientes.
- El corazón normal.
El corazón es un músculo especial que se contrae de forma regular y continua, impulsando la sangre hacia el resto del cuerpo y los pulmones. Está formado por cuatro cámaras - dos situadas en la parte superior (las aurículas) y dos en la parte inferior (los ventrículos). La contracción cardiaca se produce como consecuencia de la presencia de un flujo eléctrico que provoca dicha contracción. Estas señales eléctricas se repiten de forma cíclica y cada impulso eléctrico va a generar un latido.
- ¿Qué es la miocardiopatía espongiforme?
Se denomina miocardiopatía espongiforme (MCE) o miocardiopatía no compactada al aumento de rugosidad del músculo cardiaco en su interior que se asocia además a otras alteraciones.
La cara interna del ventrículo izquierdo es rugosa habitualmente, en todas las personas. Pero en algunas, esta rugosidad es más llamativa, dando un aspecto de esponja, por lo cual recibe este nombre.
Esta no es una enfermedad nueva aunque pudiera parecerlo. Lo que ocurre es que ha comenzado a verse esta característica en el momento en que las técnicas de imagen cardiaca han ido desarrollándose. Como las cámaras de fotos de los móviles, los equipos de ecocadiografía han mejorado la definición y la resonancia cardiaca es más accesible.
- ¿Cuánto de rugosidad se considera anormal?
Hay varias maneras de medir la rugosidad de interior del ventrículo izquierdo. Se considera anormal cuando el grosor de la capa no compactada es más del doble del grosor de la capa compactada.
- ¿Cuándo se considera el aumento de la rugosidad una enfermedad?
El hecho solo, de forma aislada, de tener un aumento de rugosidad (también llamado trabeculación) solo se considera una enfermedad si se acompaña de otras alteraciones cardiacas. Estas otras alteraciones cardiacas que pueden acompañarla son dilatación y debilidad del músculo (miocardiopatía dilatada), o aumento del grosor (miocardiopatía hipertrófica), alteraciones del electrocardiograma o arritmias cardiacas.
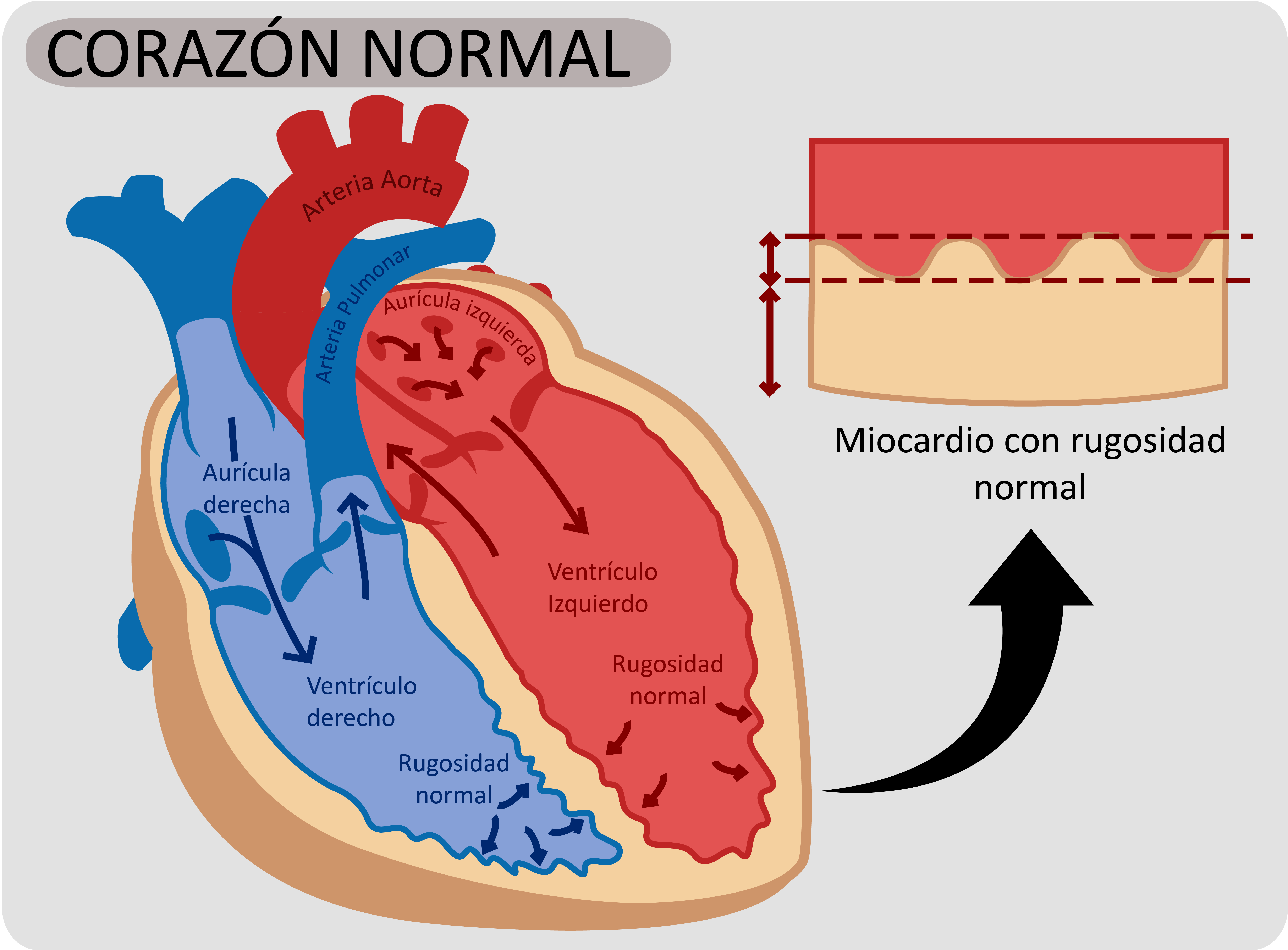

Ilustración: Eva van Passel
Rugosidad = trabeculación (son sinónimos)
Aumento de rugosidad = aumento de trabeculación = defecto de compactación = aspecto espongiforme (son sinónimos)
Cuando no hay nada más, y solo se observa el aumento de rugosidad, aunque sea llamativo, no debe considerarse una enfermedad sino una variante dentro de la normalidad. De hecho algunos cardiólogos y expertos en el tema, piensan que este aumento de la rugosidad puede ser reversible y estar en relación en algunos casos con el desarrollo de una actividad física intensa (deportista de cierto nivel) o en situaciones de mayor demanda de trabajo cardiaco, como ocurre durante el embarazo.
- ¿Conviene hacer estudios familiares y genéticos en la miocardiopatía espongiforme?
Si este aumento de rugosidad se acompañara de alguna otra alteración o hubiera una sospecha de cardiopatía hereditaria en otros miembros de la familia, entonces se recomendaría el estudio familiar. El estudio genético en los casos de aumento de rugosidad de forma aislada, aunque sea llamativa, no ofrece resultados concluyentes en este momento. El estudio genético debe solicitarse solo en aquellos casos en los que haya una enfermedad familiar clara o se identifiquen otras alteraciones.
- Prevalencia y patrón de herencia.
Alrededor de una 2% de la población tiene un aumento llamativo de la rugosidad de la cara interna del ventrículo izquierdo. En algunas personas, como los deportistas de cierta intensidad, la prevalencia puede ser superior al 5%.
En menos de 1 de cada 10 personas que tienen aumento de la rugosidad existe una enfermedad real cardiaca de base que habría que estudiar en mayor profundidad. En estos casos el estudio familiar seria aconsejable.
En los casos en los que se demuestra que existe una enfermedad familiar esta puede ser consecuencia de un defecto (mutación) en uno o más genes que puede ser transmitido entre las diferentes generaciones de una familia.
Un gen es una parte de nuestro ADN que contiene un código para la fabricación de una molécula (una proteína). La Miocardiopatía Espongiforme (MCE) puede estar causada por mutaciones en genes que codifican proteínas específicas del corazón.
Cada persona tiene dos copias de cada gen que puede estar relacionado con la MCE. Una mutación en solo una de las dos copias (que provendrá del padre o de la madre) es suficiente para desarrollar la MCE. Esto es lo que se denomina patrón de herencia autosómico dominante y el padre (o madre) que tenga la mutación tiene un 50% de riesgo de transmitir la mutación a cada hijo. Por tanto, la posibilidad de que un hijo no herede el gen mutado es también del 50%.

Patrón de herencia autosómico dominante.
En algunos casos, una nueva mutación (de novo) puede ocurrir en el ovocito, en el espermatozoide o en el embrión. En este caso, los padres del niño afecto no van a tener la mutación ni tampoco la MCE, pero el niño tiene la enfermedad y puede transmitir el gen mutado a su futura descendencia.
- Síntomas.
La mayoría de los pacientes con MCE pueden permanecer asintomáticos o estables hasta la vida adulta. Los síntomas dependen de la debilidad del corazón, en los casos en los que esta aparece, o bien por la aparición de arritmias. El hecho de tener aumento de rugosidad no es causa por si mismo de ningún síntoma. La debilidad del corazón puede ser progresiva. Un corazón débil puede tener dificultades para bombear la sangre al resto del organismo. Los síntomas más frecuentes son la falta de aire (disnea), dolor torácico, palpitaciones (debidas a arritmias), mareos y desmayos.
- Diagnóstico.
Las herramientas más comúnmente empleadas para realizar el diagnóstico de la MCE son la historia médica (tanto familiar como personal), la exploración física, el electrocardiograma (ECG), el ecocardiograma, la resonancia magnética cardiaca (RM), la ergometría o prueba de esfuerzo y la monitorización del ritmo cardiaco (Holter).
- ECG (electrocardiograma).
Este es el estudio más básico. Se colocan pequeños parches adhesivos (electrodos) en el pecho, brazos y piernas, y se conectan a través de cables con el electrocardiógrafo, que recoge la actividad eléctrica del corazón durante unos segundos. En ocasiones, es necesario realizar ECG repetidos.
- Ecocardiograma.
El ecocardiograma utiliza ondas de ultrasonidos para detectar las estructuras del corazón. Un ecocardiograma puede detectar diferentes tipos de cambios estructurales en el corazón, por ejemplo los que se producen en enfermedades como la MCE o alteraciones en las válvulas. Además, también se pueden identificar áreas de adelgazamiento de las paredes cardiacas.
- Prueba de esfuerzo o ergometría.
La prueba de esfuerzo consiste en la realización de un registro electrocardiográfico antes, durante y después de la realización de ejercicio en una cinta rodante o en una bicicleta estática. De este modo se determina la aparición de cualquier cambio en el patrón eléctrico del corazón que ocurra durante el ejercicio.
- Resonancia Magnética.
Una resonancia magnética (RM) cardiaca usa un campo electromagnético para crear imágenes del corazón. La máquina consiste en un largo tubo con una camilla en el centro que permite tumbar al paciente. La realización de la RM dura aproximadamente una hora. La RM es una técnica muy buena para mostrar la estructura de nuestro corazón, los vasos sanguíneos e identificar cualquier cicatriz (fibrosis).
- Holter (monitorización del ritmo cardiaco).
El Holter es un pequeño dispositivo digital que se coloca con un cinturón alrededor de la cintura. Además, cuatro o seis electrodos colocados en el pecho registran la actividad eléctrica del corazón durante 24-48h o hasta 7 días. Durante la monitorización todas las actividades son anotadas en un diario.
- Estudio genético.
En alrededor de la quinta parte de las familias con MCE se puede encontrar una mutación en uno de los genes que causan la enfermedad. Debido a que no todos los genes que causan MCE son conocidos, un resultado negativo en el estudio genético (es decir, cuando no se encuentra una mutación) no descarta que se trate de una causa hereditaria de MCE.
- ECG (electrocardiograma).
- Tratamiento.
Aunque no hay cura para la MCE, los tratamientos ayudan a controlar los síntomas y a disminuir los riesgos a largo plazo. La mayoría de los síntomas se controlan con medicaciones como betabloqueantes, bloqueantes de los canales del calcio, fármacos antiarrítmicos y anticoagulantes. Si los pacientes tienen un riesgo alto de muerte súbita (por ejemplo, cuando han tenido una parada cardiaca previa) o si los síntomas no pueden ser controlados con medicación, se puede implantar un desfibrilador (DAI). El DAI monitoriza de forma constante la actividad eléctrica del corazón y reconoce arritmias graves. El DAI está programado específicamente de forma individualizada para cada paciente. Puede tratar arritmias graves o rápidas enviando impulsos eléctricos o dando choques eléctricos, de modo que restaura el ritmo cardiaco normal. Un DAI está formado por dos elementos: una batería y un electrodo que monitoriza la actividad eléctrica del corazón y envía impulsos o choques eléctricos. El electrodo puede ser colocado en las cámaras derechas del corazón (a través de los vasos sanguíneos) o por debajo de la piel del tórax que cubre el corazón.
- Conclusiones respecto al diagnostico y tratamiento.
Lo cierto es que la mayoría de los pacientes con MCE, el estudio diagnóstico se basa en las pruebas de imagen cardiaca (ecocardiograma y resonancia cardiaca). Los síntomas, cuando no hay una debilidad importante, suelen ser leves y el tratamiento farmacológico consigue recuperar la debilidad del corazón y prevenir las arritmias cardiacas. Es raro que se precise de tratamientos invasivos o quirúrgicos en esta enfermedad. El riesgo de muerte súbita suele ser bajo y pocos pacientes, por tanto, necesitan un DAI.
- Estilo de vida y deportes.
Es importante reseñar que las personas que tiene aumento de rugosidad, aunque sea llamativa, que no tengan ningún otro signo de enfermedad (el electrocardiograma es normal, no tienen debilidad en el corazón y no hay arritmias en las pruebas que se realicen), no deben tener ninguna restricción para realizar una vida completamente normal. Pueden realizar ejercicio físico que deseen sin ninguna restricción.
Los que en cambio tengan ya un diagnóstico de MCE, deberían atender a algunas recomendaciones clave que ayudan a prevenir la aparición de arritmias:
- Evitar un ejercicio físico agotador- especialmente intenso, deportes competitivos.
- Realizar revisiones periódicas para monitorizar cualquier cambio.
- Recomendar a los familiares que sean examinados para detectar o descartar la presencia de la enfermedad.
El diagnóstico de MCE y la posibilidad de transmitir la enfermedad a los descendientes puede provocar ansiedad y generar una gran incertidumbre en los familiares. Los trabajadores sociales, psicólogos y especialistas médicos tienen una amplia experiencia y pueden ser de gran ayuda en este ámbito tanto para el paciente como para los familiares.
- Seguimiento.
Los pacientes en los que tras un estudio completo cardiológico lo unico que se observa es un aumento de la rugosidad cardiaca no precisan seguimiento.
En aquellos en los que se encuentran otras alteraciones cardiacas y se puede establecer el diagnóstico de MCE, el cardiólogo le aconsejará sobre la frecuencia necesaria de seguimiento, que dependerá de los síntomas, la edad y el tratamiento.
- Cribado familiar.
Si se encuentra una mutación en un gen en un paciente con MCE (ver Estudio genético), los familiares de este paciente (empezando por los familiares de primer grado: madre, padre, hermanos/as e hijos) pueden realizarse el estudio genético a través de una unidad especializada en enfermedades genéticas cardiacas. Aquellos miembros de la familia en quienes se encuentra la misma mutación (familiar) son denominados portadores y precisan un seguimiento por el cardiólogo. Aquellos otros en los que no se encuentra la mutación familiar pueden ser tranquilizados.
En caso de que no se haya identificado una mutación en un paciente con MCE, los miembros de la familia deberán ser evaluados por un cardiólogo para la realización de las diferentes pruebas diagnosticas necesarias.
La MCE puede desarrollarse a cualquier edad. Por lo tanto, se recomienda la valoración de los hijos de un afectado.
- MCE y embarazo.
Las mujeres que solo tengan un aumento de la rugosidad, aunque sea llamativa, pero que no se acompañe de ninguna otra alteración (el electrocardiograma es normal, no tienen debilidad en el corazón y no hay arritmias en las pruebas que se realicen) no se consideran enfermas cardiacas y no precisan ningún cuidado especial durante el embarazo. Durante el embarazo la rugosidad puede ser más evidente si se realiza una ecocardiografía, pero no debe acompañarse de ninguna disfunción. En cambio, las pacientes que tengan diagnóstico definitivo de MCE es importante antes de que se produzca el embarazo realizar una adecuada planificación, comentando los posibles riesgos, cambios en la medicación y los cuidados durante el mismo.

Para más información: https://guardheart.ern-net.eu



